I’ve had a couple of months off from blogging. Time for some computer-assisted biology! Robert Griffin asks on Stack Exchange about finding the distance between HP1 binding sites and genes in Drosophila melanogaster. We can get a rough idea with some public chromatin immunoprecipitation data, R and the wonderful BEDTools.
Finding some binding sites
There are indeed some ChIP-seq datasets on HP1 available. I looked up these ones from modENCODE: modENCODE_3391 and modENCODE_3392, using two different antibodies for Hp1b in 16-24 h old embroys. I’m not sure since the modENCODE site doesn’t seem to link datasets to publications, but I think this is the paper where the results are reported: A cis-regulatory map of the Drosophila genome (Nègre & al 2011).
What they’ve done, in short, is cross-linking with formaldehyde, sonicate DNA into fragments, capture fragments with either of the two antibodies and sequence those fragments. They aligned reads with Eland (Illumina’s old proprietary aligner) and called peaks (i.e. regions where there is a lot of reads, which should reflect regions bound by Hp1b) with MACS. We can download their peaks in general feature format.
I don’t know whether there is any way to make completely computation predictions of Hp1 binding sites but I doubt it.
Some data cleaning
The files are available from ftp, and for the below analysis I’ve unzipped them and called them modENCODE_3391.gff3 and modENCODE_3392.gff3. GFF is one of all those tab separated text files that people use for genomic coordinates. If you do any bioinformatics type work you will have to convert back and forth between them and I suggest bookmarking the UCSC Genome Browser Format FAQ.
Even when we trust in their analysis, some processing of files is always required. In this case, MACS sometimes outputs peaks with negative start coordinates in the beginning of a chromosome. BEDTools will have none of that, because ”malformed GFF entry at line … Start was greater than end”. In this case, it happens only at a few lines, and I decided to set those start coordinates to 1 instead.
We need a small script to solve that. As I’ve written before, any language will do, but I like R and tend to do my utility scripting in R (and bash). If the files were incredibly huge and didn’t fit in memory, we’d have to work through the files line by line or chunk by chunk. But in this case we can just read everything at once and operate on it with vectorised R commands, and then write the table again.
modENCODE_3391 <- read.table("modENCODE_3391.gff3", stringsAsFactors=F, sep="\t")
modENCODE_3392 <- read.table("modENCODE_3392.gff3", stringsAsFactors=F, sep="\t")
fix.coord <- function(gff) {
gff$V4[which(gff$V4 < 1)] <- 1
gff
}
write.gff <- function(gff, file) {
write.table(gff, file=file, row.names=F, col.names=F,
quote=F, sep="\t")
}
write.gff(fix.coord(modENCODE_3391), file="cleaned_3391.gff3")
write.gff(fix.coord(modENCODE_3392), file="cleaned_3392.gff3")
Flybase transcripts
To find the distance to genes, we need to know where the genes are. The best source is probably the annotation made by Flybase, which I downloaded from the Ensembl ftp in General transfer format (GTF, which is close enough to GFF that we don’t have to care about the differences right now).
This file contains a lot of different features. We extract the transcripts and find where the transcript model starts, taking into account whether the transcript is in the forward or reverse direction (this information is stored in columns 4, 5 and 7 of the GTF file). We store this in a new GTF file of transcript start positions, which is the one we will feed to BEDTools:
ensembl <- read.table("Drosophila_melanogaster.BDGP5.75.gtf",
stringsAsFactors=F, sep="\t")
transcript <- subset(ensembl, V3=="transcript")
transcript.start <- transcript
transcript.start$V3 <- "transcript_start"
transcript.start$V4 <- transcript.start$V5 <- ifelse(transcript.start$V7 == "+",
transcript$V4, transcript$V5)
write.gff(transcript.start, file="ensembl_transcript_start.gtf")
Finding distance with BEDTools
Time to find the closest feature to each transcript start! You could do this in R with GenomicRanges, but I like BEDTools. It’s a command line tool, and if you haven’t already you will need to download and compile it, which I recall being painless.
bedtools closest is the command that finds, for each feature in one file, the closest feature in the other file. The -a and -b flags tells BEDTools which files to operate on, and the -d flag that we also want it to output the distance. BEDTools writes output to standard out, so we use ”>” to capture it in a text file.
Here is the bash script. I put the above R code in clean_files.R and added it as an Rscript line at the beginning, so I could run it all with one file.
#!/bin/bash
Rscript clean_files.R
bedtools closest -d -a ensembl_transcript_start.gtf -b cleaned_3391.gff3 \
> closest_element_3391.txt
bedtools closest -d -a ensembl_transcript_start.gtf -b cleaned_3392.gff3 \
> closest_element_3392.txt
Some results
With the resulting file we can go back to R and ggplot2 and draw cute graphs like this, which shows the distribution of distances from transcript to Hp1b peak for protein coding and noncoding transcripts separately. Note the different y-scales (there are way more protein coding genes in the annotation) and the 10-logarithm plus one transformation on the x-axis. The plus one is to show the zeroes; BEDTools returns a distance of 0 for transcripts that overlap a Hp1b site.
closest_3391 <- read.table("~/blogg/dmel_hp1/closest_element_3391.txt", header=F, sep="\t")
library(ggplot2)
qplot(x=log10(V19 + 1), data=subset(closest_3391, V2 %in% c("protein_coding", "ncRNA"))) +
facet_wrap(~V2, scale="free_y")

Or by merging the datasets from different antibodies, we can draw this strange beauty, which pretty much tells us that the antibodies do not give the same result in terms of the closest feature. To figure out how they differ, one would have to look more closely into the genomic distribution of the peaks.
closest_3392 <- read.table("~/blogg/dmel_hp1/closest_element_3392.txt", header=F, sep="\t")
combined <- merge(closest_3391, closest_3392,
by.x=c("V1", "V2","V4", "V5", "V9"),
by.y=c("V1", "V2","V4", "V5", "V9"))
qplot(x=log10(V19.x+1), y=log10(V19.y+1), data=combined)
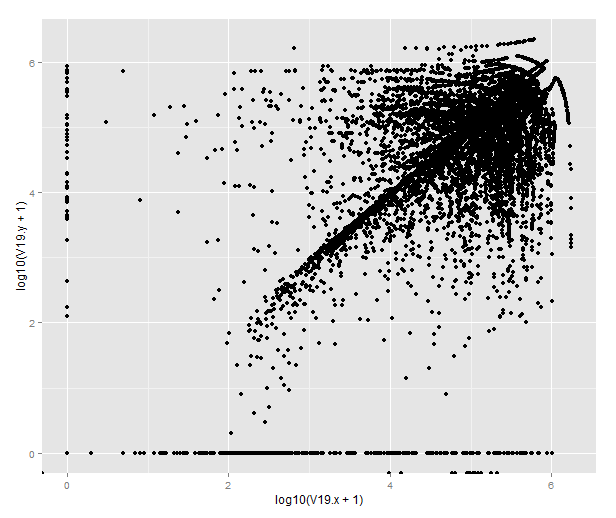
(If you’re wondering about the points that end up below 0, those are transcripts where there are no peaks called on that chromosome in one of the datasets. BEDTools returns -1 for those that lack matching features on the same chromosome and R will helpfully transform them to -Inf.)
About the DGRP
The question mentioned the DGRP. I don’t know that anyone has looked at ChIP in the DGRP lines, but wouldn’t that be fun? Quantitative genetics of DNA binding protein variation in DGRP and integration with eQTL … What one could do already, though, is take the interesting sites of Hp1 binding and overlap them with the genetic variants of the DGRP lines. I don’t know if that would tell you much — does anyone know what kind of variant would affect Hp1 binding?
Happy hacking!






